Natalia Rodón1, Olga Diaz1, Ruth Román1, Montse Verdú1,2, Yessica No Garbarino1, Isabel Trias1,2, Melina Lozano1 y Xavier Puig1,2,3.
1BIOPAT. Biopatologia Molecular SL, Grup Assistència, Barcelona; 2Histopat Laboratoris, Barcelona 3Hospital de Barcelona, SCIAS, Grup Assistència, Barcelona.
Presentamos los resultados del estudio comparativo realizado en BIOPAT para valorar la implementación de una plataforma de secuenciación masiva en sustitución de los estudios convencionales, gen a gen, para la caracterización del perfil molecular en una serie que incluye 19 carcinomas colorrectales, 26 de pulmón, 5 de páncreas y 1 melanoma, correspondientes a 51 pacientes. Se compararon los resultados de 263 genes estudiados previamente mediante técnicas convencionales con los obtenidos por secuenciación masiva (NGS). Los genes KRAS exón 2 y BRAF exón 15 se estudiaron mediante PCR alelo específica e hibridación reversa (StripAssay KRAS/BRAF), las mutaciones en los exones 3 y 4 de KRAS y 2, 3 y 4 de NRAS mediante pirosecuenciación (RasExtension PyroKit), las mutaciones de EGFR mediante real-time PCR (Therascreen EGFR RGQ), las mutaciones de PIK3CA y TP53 por secuenciación directa y los reordenamientos de los genes ALK, ROS1 y RET mediante hibridación in-situ fluorescente (FISH). El estudio de DNA mediante NGS se realizó con la plataforma Ion PGM (Thermofisher) con un kit para detectar mutaciones en 22 genes relacionados con cáncer, en una única reacción (OST DNA kit). El estudio de RNA por NGS para los reordenamientos en los genes ALK, ROS1, RET y NTRK1, se realizó únicamente en los carcinomas de pulmón (OST Fusion Kit). Cuatro de los 51 (7,8%) estudios de DNA y 4 de los 26 (15,4%) de RNA fueron no valorables mediante NGS debido a problemas técnicos que se resolvieron posteriormente.
La concordancia entre los dos estudios fue del 100% para todos los genes excepto para ROS1 (91%) debido a que en un caso se detectó una translocación de ROS1 mediante NGS que no fue detectada por FISH y, de manera inversa, en otro caso la NGS no detectó una translocación de ROS1 positiva por FISH (Tabla 1). La concordancia global entre los resultados de la serie fue del 99,2% y supone una pérdida de información del 0.4% atribuible únicamente al gen ROS1. En el 53% de los casos el estudio mediante NGS detectó mutaciones en genes no analizados anteriormente, además del caso con un reordenamiento de ROS1 no detectado por FISH, ya comentado. Con los resultados obtenidos consideramos válida la técnica de NGS en nuestro laboratorio, aunque requiere un seguimiento prospectivo con mayor volumen de casos.
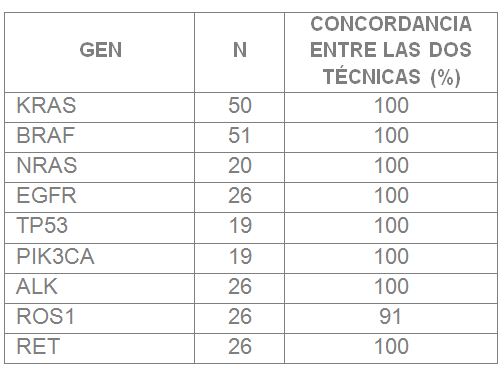
Tabla 1. Concordancia entre resultados del estudio gen a gen y NGS.